问:公司已经通过了ISO13485的认证和年度评估,办理医疗器械生产许可时药监局为什么还要来进行生产质量规范现场核查?
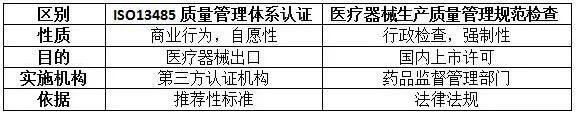
答:根据《医疗器械监督管理条例》规定,第二类、第三类医疗器械生产新开办、变更和延续均为行政许可事项,应当向所在地省、自治区、直辖市人民政府药品监督管理部门申请生产许可。《医疗器械生产质量管理规范》及附录是医疗器械生产企业必须执行的法规要求,是获得生产许可的基本条件。
ISO13485标准在国内等同转换为YY/T0287,是医疗器械质量管理体系推荐性行业标准,实施ISO13485认证的第三方认证机构不具备行政许可资格,通过第三方认证并不能作为上市许可的必要条件。
以下通过列表比较说明在我国ISO13485认证与《医疗器械生产质量管理规范》核查的区别:
问:质量部门独立行使权力在组织架构上应如何体现?有什么要求?
答:按照《医疗器械生产质量管理规范》的要求,医疗器械生产企业应建立质量管理部门,独立行使职能,对产品质量的相关事宜负有决策的权利。企业应成立专门的、独立的质量管理部门并由最高管理者或其授权代表直接管辖。质量管理部门应包括QA及QC的职能,具体职能应根据企业的实际情况在其职责文件中进行明确规定,例如质量管理、质量控制、质量检验、产品放行、让步接收、质量审核、质量数据统计分析、不合格品控制、内部审核、管理评审等。产品上市放行应由质量受权人负责,质量部门应对产品质量具有绝对的、独立的决策权,确保每批次原料、中间品、终产品符合质量要求。