2022年上海市医疗器械化妆品审评核查中心共计受理第二类有源医疗器械首次注册92件,其中91件进行了发补,发补率98.9%。下面对技术审评发补常见问题进行了分析。
一、 首次注册审评发补意见分布情况
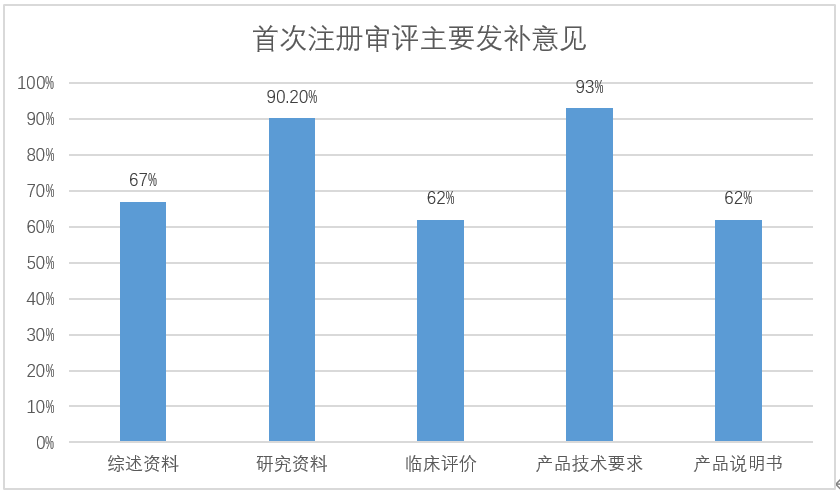
91件首次注册发补涉及68家企业,发补意见主要集中在综述资料、研究资料、临床评价、产品技术要求和产品说明书五部分(见图一),其中综述资料发补率67%,研究资料发补率90.2%,临床评价发补率62%,产品技术要求发补率93%,产品说明书发补率62%。
图一:首次注册审评主要发补意见分布情况
二、 首次注册审评常见问题分析
1、注册单元及产品管理类别问题
根据《医疗器械注册单元划分指导原则》,产品技术原理、结构组成、性能指标、适用范围不同时,原则上划分为不同注册单元。例如电子鼻咽喉镜,需要详述不同型号的差异,包括原材料、结构组成等,用于判断是否为同一注册单元。
部分产品管理类别不明确,有些产品属于三类医疗器械或不属于医疗器械,需进行分类界定,建议企业在提交资料前进行分类界定明确产品管理类别。
2、综述资料常见问题
企业应当根据《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》,编写申报产品的综述资料,存在主要问题如下:
(1)产品作用机理:未详细描述产品的工作原理,未提供产品的工作原理图,未描述功能的具体实现方式;产品原理的描述与产品检验报告中的描述不一致。
(2)结构组成:未明确具体描述产品的结构组成(包括附件、选配件);未提供各部件的详细规格;未提供内部结构示意图和电路图及说明(如,部件照片、软件的界面、功能、输出报告描述);产品结构组成与提供的产品照片中的组成不一致;未对不同型号的产品在结构组成上的差异进行详细描述。
(3)型号规格:未明确申报的产品型号和包含的部件型号之同的配置关系;对于存在多型号规格(配置)的产品,未明确产品型号划分、未明确各型号规格的区别和差异(如:结构、功能、性能指标等);对申报产品中在结构特征上有明显区别的产品,企业未在产品型号命名规则上予以区分。
(4)适用范围:未明确适用范围;产品适用范围超出《医疗器械分类目录》中对于相应分类的描述;产品适用范围的表述与配用主机的适用范围表述不一致;特定产品适用范围的内容中未明确适用人群的范围,如年龄、体重等要求。
(5)软件相关:软件功能表述不清晰,相关功能的临床用途和意义不明确;未按照软件指导原则规范制定软件发布版本与版本软件命名规则;未明确软件算法、数学模型的原理和临床依据,包括计算分析处理过程等内容;软件描述文档未按照《医疗器械软件注册审查技术指导原则》(2022年修订版)进行完善;未详细描述软件所有的交付内容和形式;未对软件安装和部署方式进行介绍;未详细描述软件架构(如BS、CS)的实现方式;未对软件中使用的现成软件进行介绍。
3、研究资料常见问题
(1)性能研究:提供的产品性能指标、产品技术要求的编制说明的确定依据不清晰不明确;强制性标准内容引用不完整,对不适用条款未给出不适用的理由;例如心电记录仪未详细说明产品对强制性标准YY0885各条款的符合性,未对不适用条款给出理由。
(2)软件研究:应按照《医疗器械软件注册审查指导原则(2022年修订版)》《医疗器械网络安全注册审查指导原则(2022年修订版)》提交产品软件研究资料和网络安全资料,存在主要问题如下:①未对软件安全级别进行详细描述,安全级别判定理由不合理,未根据软件安全性级别提交相应资料;②软件核心功能、核心算法描述过于简单,缺少测量准确性研究和验证资料;③软件描述文档中软件版本信息与检验报告不一致。④错误认定产品不含软件或不涉及网络安全。
(3)生物学研究:研究存在的主要问题有:未考虑微生物性能,未明确与患者及使用者接触部件的材料性质等。
(4)有效期和包装研究:存在的主要问题有:未根据产品特点合理选择使用期限评价路径,评价方法不清晰;有效期研究资料中未见部分不可更换部件使用寿命相关资料。
4、临床评价资料常见问题
(1)免临床评价目录(以下简称目录)内产品:与境内已注册产品的对比不全面,未考虑主要性能指标、软件功能等方面差异;对工作原理、结构组成、实现方式等显著性差异分析不充分,不能证明两者具有基本等同性;缺少对比产品注册批件或说明书等支持性资料。
(2)通过同品种器械开展评价:如提交的同品种产品临床数据欠缺可比性和充分性,未充分考虑适用范围、技术特征以及生物学特性的差异,不能证明两者具有广泛相似性;提交的临床文献中缺少同品种产品信息,未说明两者相关性;临床文献无实质内容,也未开展对比测试或验证;未对文献数据进行分析,也未评估其对申报产品性能与安全性论证的贡献。
(3)通过临床试验开展评价:如临床试验过程中对申报器械的使用操作与产品说明书不完全一致,未给出解释;适用范围描述有待规范,临床试验评价指标与企业主张的临床适用范围有显著差异;有效性评价采用评分表形式,但未提供确立依据;未对剔除病例、离群值进行合理分析;适应症表述不清晰不明确,未阐述不同病种病例数的统计学考虑。
5、产品技术要求常见问题
产品技术要求遵循的原则有《医疗器械产品技术要求编写指导原则》、适用的强制性国家/行业标准、推荐性国家/行业标准(推荐采用)。产品技术要求存在问题:
(1)缺少部分性能指标、指标不合理;
(2)缺少软件功能,软件功能描述不清晰;
(3)软件版本、命名规则不完整、缺少典型运行环境;
(4)附录中缺少必要的内容,如缺少非标体模,工装、测试软件等信息;
(5)检验方法不合理,不明确;
(6)编写不规范,如缺少标准年代号、未按最新版《医疗器械产品技术要求编写指导原则》编写;
(7)引用标准的问题,如标准条款引用不全,未识别出产品适用的标准;
(8)结构组成不完整、型号规格划分不清晰。
6、产品说明书常见问题
医疗器械产品说明书可参考以下具体文件《医疗器械说明书和标签管理规定》、标准(强标、专标)及注册技术审查指导原则对说明书的要求。常见问题如下:
(1)说明书中描述内容与综述资料、研究资料不一致;
(2)说明书中性能指标与产品技术要求不一致;
(3)未涵盖《医疗器械说明书和标签管理规定》中要求的内容或描述不清晰。
(4)说明书内容未在技术要求中体现
(5)未涵盖注册技术审查指导原则及国标行标中要求的内容。
7、注册检验报告常见问题
送检样品应为成品,检验报告上条款应与产品技术要求一致,并在具备医疗器械检验资质的检验机构进行注册检验。常见问题如下:
(1)送检样品不能覆盖所有型号规格,除选择典型型号进行全性能检验外,还应选择其他型号进行差异性检验;
(2)提交的经检测机构盖章的产品技术要求与注册提交的产品技术要求不一致。
(3)以注册人形式申报的产品,检验报告中的制造商与申报资料不一致。
三、 对本市二类有源医疗器械首次注册的建议
本文对2022年上海市第二类有源医疗器械首次注册常见发补问题进行了梳理与分析,其中最常见的问题为:产品技术要求、研究资料、综述资料。
在产品技术要求部分,应按照《医疗器械产品技术要求编写指导原则》撰写产品技术要求,注重产品技术要求编写的规范性。医疗器械产品应当符合医疗器械强制性国家标准和强制性行业标准。产品技术要求中包含的性能指标要完善,并覆盖产品的功能,否则可能会涉及补检。
关于研究资料,建议企业根据《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》进行编写,根据申报产品适用范围和技术特征,提供研究综述,逐项描述所开展的研究,提供相应的研究资料。研究资料应当包括产品性能指标的确定依据、设计输入来源等内容。研究资料应当足够充分,能够证明产品的安全性和有效性。特别对于包含软件组件的产品,应当根据《医疗器械软件注册审查指导原则(2022年修订版)》的要求提交产品软件研究资料,如涉及网络安全,还应根据《医疗器械网络安全注册审查指导原则(2022年修订版)》的要求提供相应网络安全研究资料。
综述资料部分,企业应当详细描述产品的工作原理、结构组成、适用范围、功能的具体实现方式等内容,让审评人员对产品能有整体的把握和了解,使产品的风险能更清晰地被识别。
关于说明书和标签,企业应认真研读《医疗器械说明书和标签管理规定》,按规定编制说明书和标签,规范表述产品规格型号、适用范围、适用人群、注意事项和禁忌症等。说明书与研究资料、产品技术要求等其他申报资料的内容要保持一致。
|