为帮助医疗器械注册申请人更好地理解法规、规章、指导原则等配套文件的要求,小编统计了2022年度上海市第二类无源医疗器械技术审评发补中的常见问题,并进行了归类与剖析,以帮助注册申请人提高注册申报资料的科学性与合规性,提高注册申报的质量和效率。
一、2022年上海市第二类无源医疗器械常见问题发补统计
2022年度,上海市器审中心共受理无源产品注册申报共249件。其中首次注册发补率为100%,变更注册发补率为43.47%,延续注册发补率为33.7%。
按照《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(2021年第121号)的申报资料架构,小编将发补内容进行了归类和整理。首次发布内容的发补率见下表。
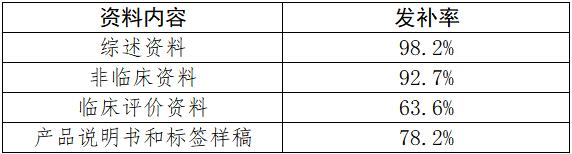
二、2022年上海市第二类无源器械首次注册常见发补问题分析
(一)综述资料常见发补问题
1.注册单元
根据《医疗器械注册单元划分指导原则》的规定,产品的技术原理、结构组成、性能指标、适用范围不同时,原则上划分为不同注册单元。例如,“液体敷料”存在西林瓶和预灌装注射器两种包装形式,产品在结构组成、使用方法存在较大差异,建议作为不同的注册单元进行注册申报。
2.产品名称
产品名称不符合《医疗器械通用名称命名规则》和《医疗器械通用名称命名指导原则》。例如,“血栓抽吸连接管”应修改为“一次性使用吸引连接管”。
3.产品作用机理
产品的作用原理描述含糊不清,未提供详细的作用机理说明或是各部件在产品中的功能、各组件连接方式。例如,“导引鞘组”的各组件如何配合以实现产品的预期用途,建议给出产品原理图和清晰示意图。
4.适用范围
产品的适用范围不规范,未能根据分类目录、分类界定文件、免临床评价目录、已上市同类产品和产品临床使用情况,规范产品适用范围。例如,“负压吸引连接管”与负压吸引泵配合使用,根据临床实际使用在适用范围中明确配套使用的负压源情况,给出产品能够承受的最大负压值。
5.结构组成
产品的结构组成不明确,未能提供清晰示意图。建议明确产品的具体结构,提交产品示意图,标注主要尺寸和组件名称以及产品各组成部分的清晰图片。
6.型号规格
产品型号规格的设定依据不足、不同规格之间的划分不明确,不同型号规格之间的差异描述不清楚。例如,“润滑剂”规格从5mL到1000mL,需要补充提交临床应用依据,建议采用对比产品表对不同规格型号的结构组成、功能、性能指标加以描述。
7.原材料
缺少产品原材料符合相关标准的支持性资料。建议提供每个组件的原材料详细信息,包括原材料的等级规格、质量标准、供应商和入厂检验要求;若生产过程中含有溶剂、粘结剂、添加剂等,应进一步明确。原材料如为液体,建议提供配比依据,不同配方可能影响产品的基本性能。
(二)非临床资料常见发补问题
1.产品技术要求
型号规格常见问题主要是:未明确型号规格划分依据。性能指标发补问题有:①产品技术要求中的产品性能指标低于适用的国家标准/行业标准;②未采用最新发布的国家标准、行业标准;③与国行标不一致或有缺漏适用项;④检验方法与国家/行业标准不一致且未提交充分验证依据。
2.检验报告
检验报告的典型性覆盖问题较为常见,如送检样品未能覆盖全部的型号规格,建议除选择典型型号进行全性能检验外,还应选择其他型号进行差异性能检验。
3.性能研究
①申请人往往对于产品技术要求中的性能指标的研究,仅提供了部分化学和物理性能编制研究资料。例如,企业提供了性能指标的确定依据,但未提供性能验证方案和报告;②申请人未能提供对于产品安全性、有效性存在一定影响,但未纳入产品技术要求的性能指标中的其他性能研究资料,例如:“一次性使用导引鞘”除产品技术要求中的性能指标外,还应补充提交模拟使用相关的研究资料,如推送性能、扭转性能、回撤性能、抗弯折性能、抗扭结性能等;
4.生物相容性研究
①产品的生物相容性研究评价依据、接触时间和与人体接触的性质等不明确,如仅提供原材料生物学试验报告,并非成品的生物学试验报告;②试验样品不具有代表性,如鼻塞导管仅选用成人型为代表型号,该型号未覆盖婴儿型号的胶体贴片(直接接触婴儿皮肤)。
5.灭菌研究
①提交的灭菌研究资料不完整,如:企业未能根据GB 18279系列标准提交灭菌确认方案和报告、环氧乙烷的解析验证方案及报告、相关过程记录附件;②所选灭菌产品不能代表注册单元内的所有规格型号;③企业未能详细描述过程挑战装置(IPCD和EPCD)的制备方法、明确BI的放置位置及最难灭菌位置的确定依据;④关于灭菌产品的追加,应根据GB 18279系列标准和YY/T 1268-2015《环氧乙烷灭菌的产品追加和过程等效》评估灭菌确认的产品是否能列入同一灭菌处理组,并提供论证性资料,必要时应对灭菌组内的所有产品及其内部挑战装置进行短周期抗性对比试验以确认最难灭菌的产品;不同灭菌柜也应提交等效性验证资料;⑤产品经全周期灭菌后应对产品性能和包装性能进行检测。如:企业未检测灭菌后产品和包装的全性能指标。
6.有效期和包装研究
①应依据《无菌医疗器械包装试验方法第1部分:加速老化试验指南》(YY/T 0681.1)对产品(包括包装)采用加速老化试验和实时老化试验的方式验证其有效期;②老化验证报告应包括但不限于,加速老化起止时间、加速老化设备、老化产品批号,并明确产品零时刻的检验报告和终时刻的检验报告;③进行有效期验证的产品应能代表注册单元内的所有型号规格;④产品有效期验证的检测性能应包含产品技术要求中的所有性能和包装性能,明确性能指标和检测方法,提交测试记录,并对加速老化过程中产品性能的变化进行分析。⑤产品包装验证可依据有关标准进行(如GB/T 19633、YY/T 0681.1等),提交产品的包装验证报告。直接接触产品的包装材料的选择应至少考虑以下因素:包装材料的物理化学性能;包装材料的毒理学特性、包装材料与产品的适应性、包装材料与成型和密封过程的适应性、包装材料与标签系统的适应性、包装材料与贮存运输过程的适应性。
(三)临床评价资料常见发补问题
1.免临床评价目录
根据《免于临床评价医疗器械目录》内容,注册申请人应提交申报产品与目录产品的对比资料,包括结构组成、材质、适用范围等信息。
2.通过同品种临床评价路径进行临床评价
对于与境内已上市产品的对比项目中,应对其结构差异、型号规格、预期用途等进行详细的分析评价,论证存在的差异是否会产生不同的安全有效性问题,并提供科学依据和支持性资料。同时,建议注册申报人的比对资料详细完整,不要使用“无明显差异”“基本等同”等模糊描述,对差异部分应进行详细论述。
(四)产品说明书和标签样稿常见发补问题
医疗器械产品说明书可参考以下具体文件《医疗器械说明书和标签管理规定》、标准(强标、专标)及注册技术审查指导原则对说明书的要求。常见发补问题如下:说明书中性能指标与产品技术要求不一致;说明书中描述内容、适用范围与综述资料、研究资料不一致;未涵盖《医疗器械说明书和标签管理规定》中要求的内容或描述不清晰。
三、上海市2022年第二类无源器械变更注册常见发补问题分析
变更注册最常见发补问题是对变更内容的验证和确认不够充分,或表述不规范。大部分问题与前文首次注册常见问题相同,不再赘述。变更注册特有的发补内容如下:
(一)综述资料常见发补问题
企业应根据产品具体变更情况提供相应的说明及对比表:型号规格变化、产品名称变化、产品技术要求变化、结构及组成变化、产品适用范围变化等。特别注意如果新增型号规格与原有型号规格在结构组成、性能指标、适用范围等方面有显著差异,不能作为同一注册单元进行变更注册。
(二)研究资料常见发补问题
涉及新增型号规格,需提交临床设计的依据,及相应的性能研究、灭菌研究、有效期研究、临床评价等方面的资料;涉及部件材料发生变化,需提交成品性能研究报告、有效期验证方案和报告,必要时提供生物学评价报告;涉及变更灭菌方式,应提供生物相容性研究资料,以评估灭菌方式的改变是否会影响产品的生物相容性。
四、上海市2022年第二类无源器械延续注册常见发补问题分析
延续注册发补最主要问题是执行的强制性标准、《中华人民共和国药典》版本已修订,但申请人未在产品技术要求中补充新要求并提供证明产品符合新要求的检验报告。该情况下,会在形式审查阶段发出补正意见。申请人应持续关注新的强制性标准发布情况,确保相关条款符合国家标准要求后再行延续注册申报。
五、总结与讨论
本文对2022年上海市第二类体无源器械注册申报常见发补问题进行了系统的梳理和分析。无源产品种类繁多,确认和验证的过程较复杂,对申请人能力要求较高。申请人应深刻理解、准确把握相关指导原则中的具体要求,再实施验证和确认,避免发补后重新进行验证,造成时间与资源的浪费。产品技术要求、产品说明书、文字描述的规范性、与注册申报资料的一致性应高度重视,仔细斟酌。由于新发布的《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告(2021年第121号)》对综述资料有了更具体的要求,申请人应予以相应关注,避免出现资料完整性的问题。
|
