一、独立软件现场检查概况
依据《医疗器械生产质量管理规范》(以下简称《规范》)和《医疗器械生产质量管理规范附录独立软件》(以下简称《软件附录》),2023年上海器审中心对本市30家医疗器械软件企业开展了38次现场体系核查,其中包括35次注册质量管理体系核查和3次生产许可现场体系核查。注册核查涉及37个独立软件产品,其中二类产品32个、三类产品5个。相比2022年,核查企业数量增加约67%,涉及独立软件产品数量增加80%,涉及的产品种类主要为医学图像处理类、体外诊断类、医学数据处理类等。
二、不符合项分布情况
因疫情结束及产品申报数量增加,2023年度独立软件现场核查不合格项数量相比2022年大幅增加,以下括号内为2022年数据。不符合项合计302项(143项),其中重点项60项(27项),一般项242项(116项)。单次检查发现不符合项最多为18项,最少为1项,其中重点项最多为5项,最少为0项(见图1)。
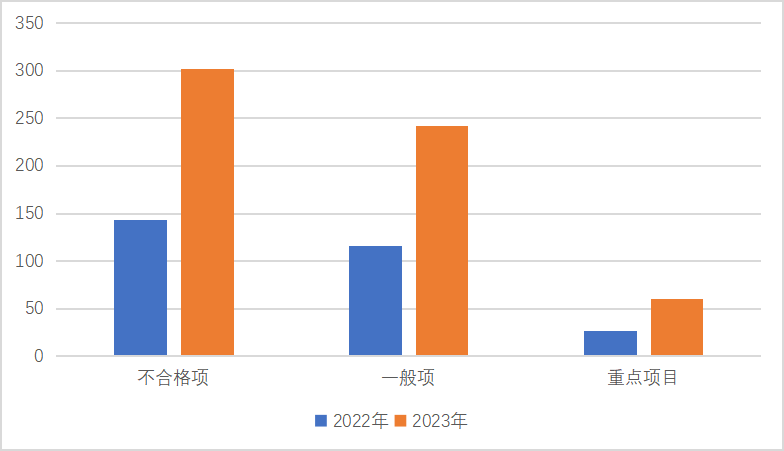
图1:2022年与2023年核查不合格数对比
从不符合项在规范独立软件附录各章节的分布情况来看,设计开发占比最多,较2022年更集中。占前五位的分别为设计开发(75%)、生产管理(6%)、机构与人员(4%)、质量管理(4%)和文件与记录(3%)。2022年占前五位的分别为设计开发(59%)、生产管理(8%)、机构与人员(8%)、质量管理(6%)和文件与记录(5%)。
进一步分析设计开发部分,主要不合格项分布在“软件测试(18.1%)、需求分析(10.6%)、版本控制(8.8%)、缺陷管理(8.8%)、配置管理(8.4%)等五个方面。
三、现场核查存在问题
通过汇总及分析,在《规范》实施过程中,企业主要存在以下方面问题:
(一)软件测试记录不完整,软件单元测试、集成测试以及系统测试未根据测试计划实施,未及时形成相应软件完整的测试记录、报告以及评审记录,并适时更新;同时软件测试用例过于简单,未覆盖风险项,可操作性差,测试数据使用不规范,导致验证不充分。
(二)软件需求不明确,首先是内容不完整,未覆盖独立软件产品所有功能,其次描述不具体,缺乏对功能、性能、以及标准等相关内容详细描述,导致设计人员无法理解真实需求。
(三)软件版本变更不规范,未按软件版本命名规则的进行管理变更,部分企业存在双重标准。
(四)软件缺陷管理不规范,缺乏缺陷分析评估、软件缺陷修复、回归测试、风险管理、配置管理、评审等活动的相关要求及记录。
(五)软件配置管理不完整,软件配置管理要求不明确,配置管理记录不完整。
|
